
Nouvelle publication de documents confidentiels de Pfizer, « 10 000 pages de données sur les vaccins de Pfizer-BioNTech »

Le 1er mars, la nouvelle tranche tant attendue des documents de Pfizer a été rendue publique grâce à la récente décision judiciaire. 10 000 pages d’une cache de plus de 450 000 données relatives aux vaccins Pfizer-BioNTech, sur lesquelles la FDA s’est appuyée pour accorder l’autorisation d’utilisation d’urgence, peuvent désormais être consultées.
La première vague de documents a été publiée en novembre dernier, à la suite d’une demande d’accès à l’information présentée par le groupe de plaignants, Public Health and Medical Professionals for Transparency (PHMPT), composé de plus de 30 scientifiques, professionnels de la santé et universitaires, dirigé par le Dr Peter McCullough et représenté par Aaron Siri, du cabinet Siri & Glimstad LLP.
En décembre dernier, j’ai rédigé un rapport d’enquête pour TrialSite News sur l’analyse cumulative des effets indésirables des vaccins par Pfizer, un document choquant de 38 pages qui faisait partie de la première vague de dossiers publiés. Le document révèle que plus de 1 228 décès sont survenus après l’administration du vaccin Pfizer BioNTech et que 42 086 personnes (cas) ont signalé 158 893 effets indésirables du vaccin, dont beaucoup étaient graves, sur une période de trois mois.
Jusqu’en janvier, la FDA a mené une bataille juridique pour empêcher la publication de ces données, en violation de la loi sur la liberté d’information. L’agence « traînait les pieds » et n’était disposée à produire que 500 pages par mois – ce qui signifie que le public devrait attendre 75 ans pour voir tous les documents. Le 6 janvier, le juge Mark Pittman a ordonné à la FDA de rendre publics tous les documents de Pfizer dans un délai de 8 mois, à raison de 55 000 pages par mois.
Voici un résumé de mes conclusions après un premier examen de la pléthore de documents en un temps limité.
Les formulaires d’exposé de cas (CRF)
Un Case Report Form (CRF) est un document imprimé ou électronique utilisé dans la recherche sur les essais cliniques pour saisir les données cliniques standardisées de chaque patient, y compris les événements indésirables. Il s’agit d’une partie essentielle du processus d’essai clinique et joue un rôle important dans la pharmacovigilance.
La majorité des CRF publiés provenaient de divers sites d’essais gérés par Ventavia, l’un des groupes de recherche clinique engagés par Pfizer pour mener les essais du vaccin Covid-19. L’entreprise fait actuellement l’objet d’un procès intenté par Brook Jackson, l’ancienne directrice régionale de Ventavia, devenue lanceuse d’alerte, qui a fourni au BMJ une prépondérance de documents et de photos internes à l’entreprise, révélant la mauvaise gestion des laboratoires du contractant de Pfizer et la compromission de l’intégrité des données et de la sécurité des patients. Mme Jackson s’entretiendra en exclusivité avec TrialSite News dans une prochaine interview sur cette affaire. Les lecteurs se souviendront peut-être que Facebook a littéralement vérifié les faits dans le BMJ pour le reportage sur cet incident. Ils n’avaient aucune raison de censurer l’article de la revue médicale indiquant la possibilité d’un biais algorithmique programmatique.
Les erreurs et anomalies
Le sujet n° 11281009 faisait partie des essais de phase 2/3 de Pfizer dans la population saine. Cette cohorte a été jugée éligible par le jugement clinique de l’investigateur en répondant aux critères de « santé ».

L’examen des antécédents médicaux généraux de ce participant montre qu’il était loin d’être en bonne santé. Le participant était un diabétique de type 2, souffrait d’angine de poitrine et s’était fait poser un stent cardiaque après un infarctus du myocarde (crise cardiaque).
Il est difficile de comprendre comment un investigateur de Ventavia a pu considérer ce participant comme sain et l’inclure dans l’essai. J’ai rencontré d’autres participants qui ont été inclus dans ces phases d’essais (sur la population en bonne santé) et dont les antécédents médicaux généraux comportaient une longue liste d’affections. Quelle pression le promoteur (Pfizer) a-t-il exercée sur l’organisme de recherche sous contrat et les sites d’essai participants pour le recrutement des participants à l’essai du vaccin ?

Un autre CRF pour ce participant révèle un événement indésirable d’infarctus du myocarde (crise cardiaque) nécessitant une hospitalisation, noté comme grave ; cependant, le numéro de l’événement indésirable grave (EIG) a été laissé en blanc (voir la capture d’écran ci-dessous). Plus tard, un numéro d’EIG a été saisi, mais il est surprenant que les associés de recherche clinique fassent des erreurs de déclaration de données aussi importantes que celle-ci. Les numéros d’EIG laissés en blanc étaient-ils courants sur les sites d’essai de Ventavia ? Encore une fois, à quel type de pression les ORC et les sites étaient-ils exposés ?

Un autre point digne d’intérêt est la date de début et de fin de ces EIG. La date de début de l’infarctus du myocarde est enregistrée le 27 octobre et la date de fin le jour suivant, qui se trouve être la date de début de la pneumonie (voir la capture d’écran ci-dessous).

Il est intéressant de noter que l’issue de l’infarctus du myocarde est enregistrée comme « rétablie/résolue » (voir la capture d’écran ci-dessous) et que la date de fin saisie n’est enregistrée qu’un jour après la date de début. Cette anomalie soulève des doutes quant à l’exactitude de ces dates enregistrées, ce qui pourrait constituer une violation des directives ALOCA-C relatives à la documentation des sites cliniques pour les essais cliniques. C’est-à-dire que les données doivent être :
- Attribuables
- Lisibles
- Contemporaines
- Originales
- Précises
- Complet

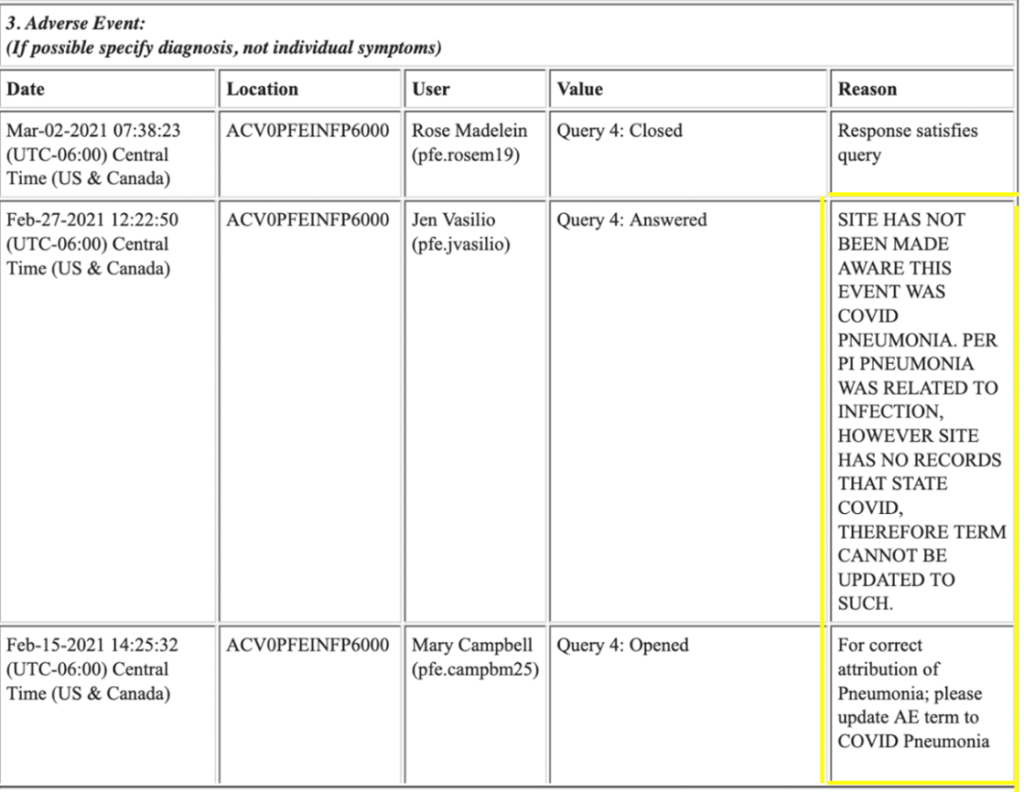
En ce qui concerne l’EIG de la pneumonie, nous pouvons voir ci-dessous que l’investigateur de l’essai, Salim Boguermouth, a entré « une pneumonie potentielle liée à COVID-19 aurait dû déclencher une visite de la maladie de Covid ». Le fait qu’il s’agisse d’une question ouverte prouve que le protocole n’a pas été suivi de manière cohérente.

Un autre enquêteur ouvre la même requête, déclarant que le terme AE de pneumonie devrait être mis à jour en Pneumonie Covid. La réponse est intéressante car elle indique simplement que « le site n’a pas été informé qu’il s’agissait d’une pneumonie Covid. Selon le PI (principal investigateur), la pneumonie est liée à une infection, le terme ne peut donc pas être mis à jour en tant que tel ». Cette réponse semble satisfaire la demande et elle est close. Aucune autre question n’a été posée ; aucune enquête ne semble avoir été effectuée. (Voir la capture d’écran ci-dessous)
Dans le protocole de Pfizer (section 8.2.4), le COVID-19 amélioré (renforcement dépendant des anticorps potentiellement causé par le vaccin) figurait sur leur liste de surveillance, ce qui indique qu’ils étaient préoccupés par cette affection. Il est important de noter que les équipes non aveugles examinaient les cas de COVID-19 sévère et les EI pour d’autres cas potentiels.
Dans la phase 2/3, l’équipe non aveugle soutenant le DMC, y compris un moniteur médical non aveugle, examinera les cas de COVID-19 sévère au fur et à mesure qu’ils seront reçus et examinera les EI au moins une fois par semaine pour détecter d’autres cas potentiels de COVID-19 sévère. A tout moment, l’équipe non aveugle peut discuter avec le président du CGD pour savoir si le CGD doit examiner les cas de déséquilibre défavorable des cas de COVID-19 et/ou de COVID-19 sévère entre le groupe vacciné et le groupe placebo ».
Par inadvertance, cela aurait pu entraîner un biais, car les équipes sans insu auraient su quels participants avaient reçu le placebo et ceux qui avaient reçu le vaccin. Elles auraient pu subir des pressions de la part du promoteur pour que l’essai se déroule d’une certaine manière et que des événements comme la « pneumonie de Covid » soient classés simplement comme une pneumonie.
Étant donné que les directives non contraignantes de la FDA aux fabricants de vaccins contre le covid-19 leur demandent de trouver une méthode pour permettre aux volontaires du bras placebo de leurs études de recevoir le vaccin, en octobre 2020, les participants à l’essai de Pfizer assignés au placebo se sont vus offrir le vaccin.
Cela aurait déclenché la levée de l’aveuglement du participant et de toutes les autres personnes impliquées. Étant donné que près de la moitié des participants auraient reçu le placebo au cours des phases 1/2/3 des essais, il est juste de dire qu’une partie importante d’entre eux auraient été évalués comme éligibles pour le véritable vaccin. Les données recueillies sur ces participants n’auraient pas été soumises à l’insu. Cela soulève une question importante : les études sans insu (observationnelles), par opposition aux études à double insu (où le participant et les personnes administrant le traitement sont en aveugle), sont sujettes à des biais importants qui peuvent affecter de manière significative l’intégrité des données.
Une étude d’examen systématique a été menée et publiée dans l’International Journal of Epidemiology, dans ses conclusions, il est dit : Cette étude fournit des preuves empiriques d’un biais prononcé dû à l’absence d’aveuglement des patients dans les essais cliniques randomisés sur les médecines complémentaires/alternatives avec des résultats rapportés par les patients.
Cependant, selon le protocole d’essai clinique de Pfizer, ses essais (qui sont toujours en cours) ne sont pas réalisés en double aveugle mais en « aveugle observateur », c’est-à-dire que le personnel du promoteur, les directeurs d’étude, les associés de recherche clinique et les personnes chargées de « garantir les exigences du protocole » ne sont pas en aveugle.

En levant l’insu sur les essais de vaccins pour ce qu’au moins certains experts considèrent comme un nouveau produit de thérapie génique, Pfizer a-t-il créé un nouveau précédent ? Dans une interview accordée au British Medical Journal (BMJ), Steven Goodman, doyen associé de la recherche clinique et translationnelle de l’université de Stanford, a déclaré qu' »en autorisant la levée de l’insu, on établit une norme de facto pour tous les essais de vaccins à venir, ce qui est dangereux ».
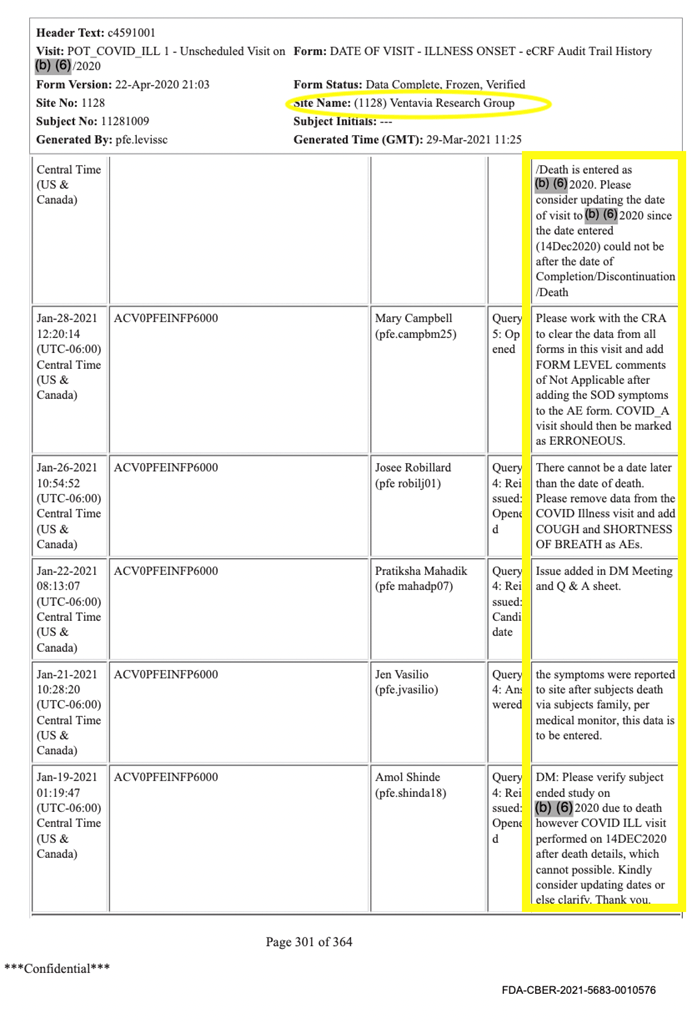
L’une des erreurs et anomalies les plus significatives trouvées sur les CRF du sujet #11281009 est peut-être celle ci-dessous, qui révèle étonnamment que le décès du participant a été enregistré avant une visite « Covid ill ». Bien entendu, il est impossible qu’un sujet décède puis se rende à l’essai clinique et y participe.
L’investigateur clinique en prend note en écrivant : « Il ne peut y avoir de date postérieure à la date du décès. Veuillez supprimer les données de la visite de maladie COVID et ajouter la toux et l’essoufflement en tant qu’EI (événements indésirables) ». Quel genre de pression était exercé ici ?
Sujet n° 11281014
Ce participant a été inscrit sur le même site Ventavia (1085). Le participant a reçu la première dose du traitement en aveugle le 31 juillet et la deuxième dose le 27 août (en dehors du protocole de la fenêtre de 3 semaines).

La capture d’écran ci-dessus montre quand la deuxième dose a été administrée. À ce stade, l’auteur souhaite soulever une question préoccupante, étant donné que presque tous les CRF examinés à l’entrée standard de la ligne 10 comprennent le terme suivant : « La période d’observation spécifiée par le protocole » a été saisie, certains CRF indiquant « 30 minutes ». Cela fait référence à la période pendant laquelle le sujet est observé par le personnel de l’essai après avoir reçu le traitement. Il est important de noter que 30 minutes est la durée minimale pendant laquelle le sujet doit être observé après le traitement. Le fait que la majorité des CRF se contentent d’indiquer ce qui semble être une entrée automatique pour la ligne 10 est préoccupant, car on peut se demander si les participants n’ont pas été observés pendant une durée adéquate, mettant ainsi leur sécurité en danger. Cela confirme ce que Brook Jackson, le dénonciateur de Ventavia/Pfizer, a déclaré dans de nombreuses interviews.
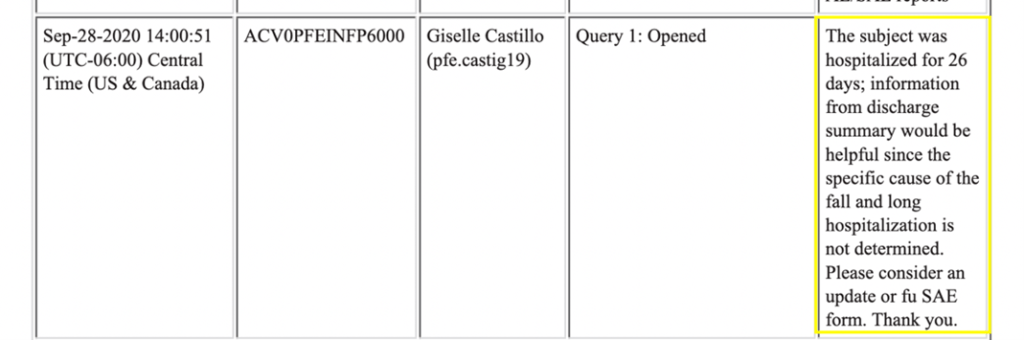
Ce qui est inhabituel dans les CRF de ce sujet, c’est qu’ils révèlent que ce participant a fait une chute grave, le lendemain, le 28 août, après l’administration de la deuxième dose, ce qui a entraîné son hospitalisation. (Voir la capture d’écran ci-dessous)

La chute a causé des lacérations faciales, qui ont été enregistrées comme un EI distinct mais n’ont pas été signalées comme graves, même si le niveau de toxicité attribué était de 2 et que le participant a été hospitalisé pendant 26 jours, voir ci-dessous.
La capture d’écran ci-dessous montre un rapport d’AE pour des lacérations faciales.

La ligne 9 comprend une anomalie inhabituelle indiquant que l’événement n’est » PAS LIÉ » au traitement de l’étude, mais qu’il s’agit d’une » hypotension « , alors que dans le formulaire de rapport d’EI pour la » chute » (voir la capture d’écran ci-dessous), l’événement est dû à une » chute « .

La capture d’écran ci-dessous montre un numéro SAE manquant pour « lacérations faciales ».

Ceci a été signalé par un investigateur de l’essai, voir ci-dessous.

Pour ces deux EIG, le personnel de Ventavia a déclaré que les deux événements étaient dus à « d’autres raisons » et n’étaient pas liés au traitement de l’étude. Cependant, on peut douter de la crédibilité de cette information étant donné que la chute et les lacérations faciales étaient intrinsèquement liées. Ainsi, si les lacérations faciales étaient dues à une « hypotension », la chute devait également être due à cette raison.
Il convient de noter que la chute s’est produite le lendemain de l’administration de la deuxième dose de traitement, ce qui soulève au moins la question de la causalité.
Il est également inquiétant que la capture d’écran ci-dessous montre comment l’ARE #2020337848 (ce numéro est référencé à la ligne 15 du rapport d’EI ci-dessus pour la chute) « la causalité a été enregistrée comme RELATIVE dans le formulaire SAE, mais a été rapportée comme NON RELATIVE sur le CRF AE ».

Sujet n° 11281103
Les antécédents médicaux généraux de cette participante ne montrent aucun signe d’altération de la fonction rénale (comme une hypokaliémie ou des calculs rénaux).
Elle a reçu la première dose du traitement en aveugle le 12 août et la deuxième dose le 1er septembre. Un mois plus tard, on signale qu’elle a des calculs rénaux, une hypokaliémie et une infection des voies urinaires le 3 octobre.

Toutes les dates de début enregistrées correspondent, ainsi que les dates de fin enregistrées.
Le rapport AE pour les calculs rénaux est ci-dessous.
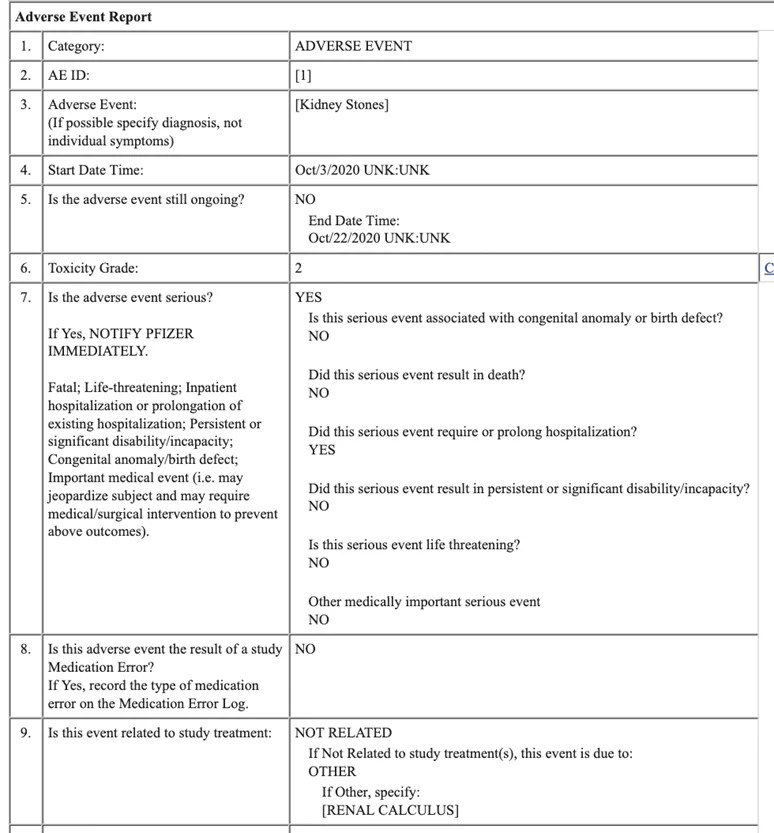
L’entrée de la ligne 9 montre que « cet événement est dû à un autre… calcul rénal » et pour l’EI d’hypokaliémie sévère (voir ci-dessous), l’événement est attribué à « l’hypokaliémie ». Les deux événements sont « NON RELIÉS » au traitement de l’étude tel que rapporté par le personnel de l’essai.

Étant donné que ce participant n’avait pas d’antécédents d’insuffisance rénale avant de prendre part à l’essai et que des calculs rénaux et une insuffisance rénale ont été signalés comme des effets secondaires du vaccin Pfizer-BioNTech, on peut se demander pourquoi ces EI n’ont pas fait l’objet d’un examen plus approfondi pour déterminer s’ils étaient liés au traitement de l’étude, en particulier lorsqu’ils sont apparus un mois seulement après la deuxième dose de traitement.

Les numéros manquants des événements indésirables graves (EIG)
En examinant les CRF du participant, sujet n° 10851246, un rapport d’EI est enregistré avec la mention « Exposition pendant la grossesse » pour l’événement indésirable. Ce terme est utilisé lorsqu’on découvre qu’une participante est enceinte pendant ou après l’arrêt de l’étude.

Une demande est faite concernant le numéro de SAE laissé en blanc pour ce participant sur un autre site Ventavia (1085).
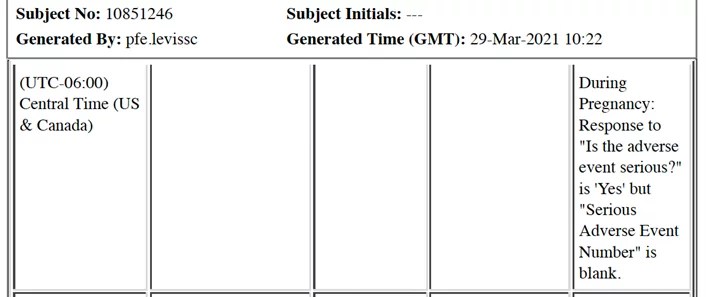
Pour le sujet n° 10851216, un numéro d’événement indésirable grave est laissé en blanc concernant une « fracture de la jambe gauche » après une chute.
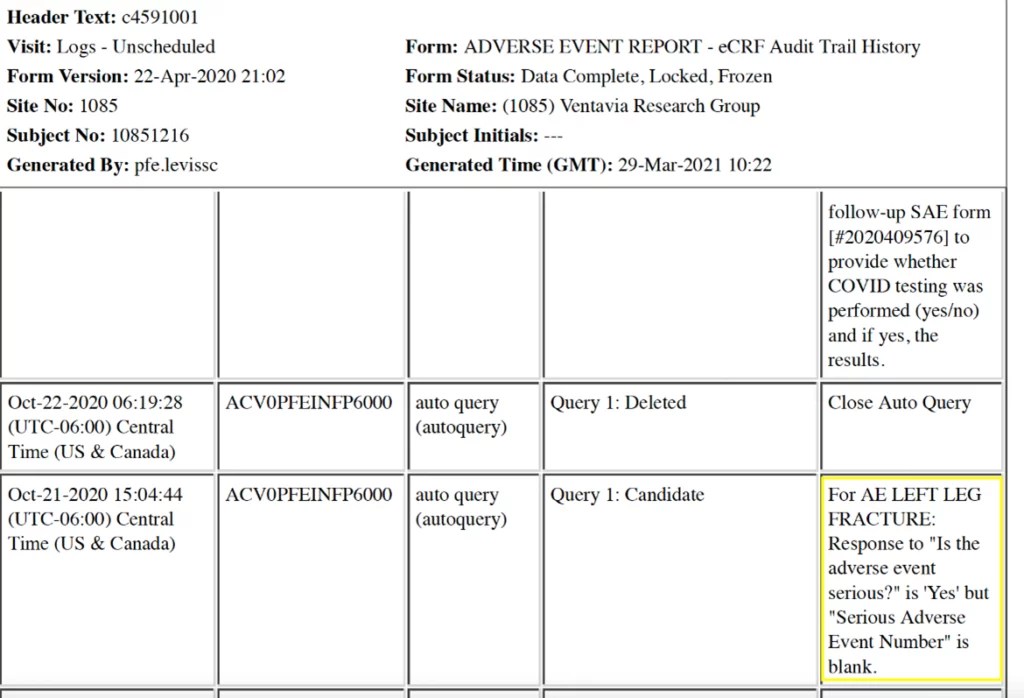
Sur le site Ventavia #1085, il semble y avoir une tendance à laisser les numéros SAE en blanc.
Les codes-barres manquants
Au cours de la rédaction de ce rapport, l’auteur a non seulement examiné des milliers de CRF, mais il a également rencontré de nombreuses entrées de codes-barres manquants pour des échantillons prélevés sur des participants, comme celui ci-dessous. Cela suggère une possibilité sérieuse que des preuves suffisantes révèlent un modèle de données douteuses sur les sites d’essai Ventavia, au mieux, peut-être compromis dans des scénarios plus graves.

Tous les éléments recueillis en peu de temps semblent confirmer les affirmations de la dénonciatrice Jackson concernant la mauvaise gestion des données sur les sites d’essais et soulèvent des questions quant à la manière dont Ventavia a mené les essais cliniques de Pfizer. Les erreurs et anomalies dans les CRF font également allusion à ses affirmations selon lesquelles les associés de recherche clinique n’étaient pas formés de manière adéquate, nombre d’entre eux n’ayant aucune expérience clinique préalable. Si des constatations aussi flagrantes sont vraies sur ces sites, pourraient-elles se manifester sur d’autres sites d’essais en Amérique du Nord et au-delà ?
Il convient de souligner que la FDA n’a effectué des inspections que dans 1 % des sites d’essais cliniques.
*
Note aux lecteurs : Veuillez cliquer sur les boutons de partage ci-dessus ou ci-dessous. Suivez-nous sur Instagram, @globalresearch_crg et sur Twitter, @crglobalization. N’hésitez pas à reposter et à partager largement les articles de Global Research.
